

Lillonum
Célébrez la Journée mondiale de l’art avec nous !

Coup de théâtre !
Crédit image : Saverio della Gatta, Performers of a Commedia dell'Arte, 1827, WikiCommons .

Projet CoESciTer
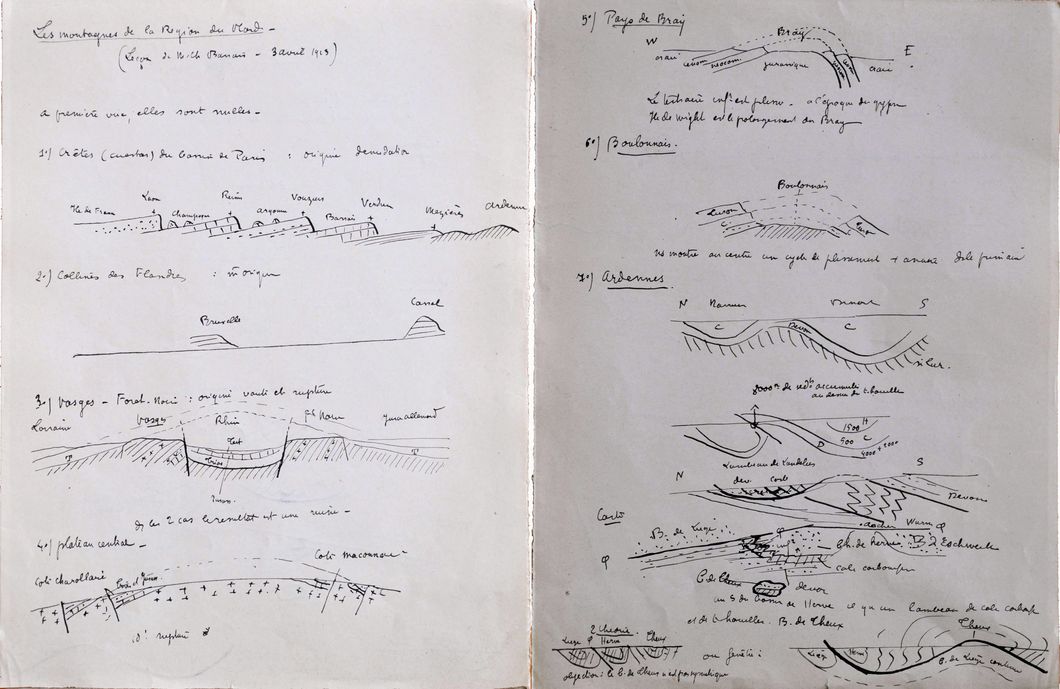
Dès que le printemps revient

LillOnum
5 803 documents disponibles dans LillOnum


Nouveautés

Opinion du Comte de Mirabeau, Sur le Réglement donné par le Roi, pour l'exécution de ses Lettres de Convocation aux prochains Etats-Généraux de son Comté de Provence...
Date : 1789
Sujet : France. États provinciaux de Provence (....-1789) France. États généraux (1789 ; Versailles)

Observations de M. Mirabeau l'ainé, relativement a l'Essai sur la proportion de l'or à l'argent qui seroit la plus convenable dans la monnoie de France, par M. F. Solignac
Date : 1790
Sujet : Bimétallisme -- France -- 1789-1799 (Révolution) Monnaie -- Frappe Monnaies d'or Monnaies d'argent

Les nouvelles galantes et comiques. Tome 3
Date : 1669

Les nouvelles galantes et comiques. Tome 2
Date : 1669

Les nouvelles galantes et comiques. Tome 1
Date : 1669

Du fanatisme dans la langue révolutionnaire ou de la persécution suscitée par les Barbares du dix-huitième siècle, contre la Religion chrétienne et ses ministres par Jean-François La Harpe
Date : 1797
Sujet : Langage politique -- France--1789-1815 Église catholique -- France -- 18e siècle France -- 1789-1799 (Révolution) -- Aspect religieux Église et État -- France -- Histoire --1789-1815

Plan de division du royaume et réglement pour son organisation, présenté par M. Le Comte de Mirabeau à l'Assemblée nationale
Date : 1789
Sujet : Administration locale -- France -- 1789-1799 (Révolution) Compétence territoriale -- France -- 1789-1799 (Révolution)

Discours d'un fidèle sujet du Roy touchant l'establissement d'une Compagnie françoise pour le commerce des Indes orientales
Date : 1665
Sujet : Compagnie royale des Indes Orientales (France ; 1664-1719) Europe -- Commerce -- Asie -- Histoire Navires de commerce -- Europe -- 1500-1800


The history and antiquities of the Exchequer of the kings of England, in two periods : to wit, from the Norman conquest, to the end of the reign of K. John; and from the end of the reign of K. John, to the end of the reign of K. Edward II. Taken from records. Together with a correct copy of the ancient dialogue concerning the exchequer, generally ascribed to Gervasius Tilburiensis. And a dissertation concerning the most ancient great roll of the exchequer, commonly styled The roll of Quinto Regis Stephani. Volume 2
Date : 1769
Sujet : Finances publiques -- Grande-Bretagne

The history and antiquities of the Exchequer of the kings of England, in two periods : to wit, from the Norman conquest, to the end of the reign of K. John; and from the end of the reign of K. John, to the end of the reign of K. Edward II. Taken from records. Together with a correct copy of the ancient dialogue concerning the exchequer, generally ascribed to Gervasius Tilburiensis. And a dissertation concerning the most ancient great roll of the exchequer, commonly styled The roll of Quinto Regis Stephani. Volume 1
Date : 1769
Sujet : Finances publiques -- Grande-Bretagne
