

Lillonum
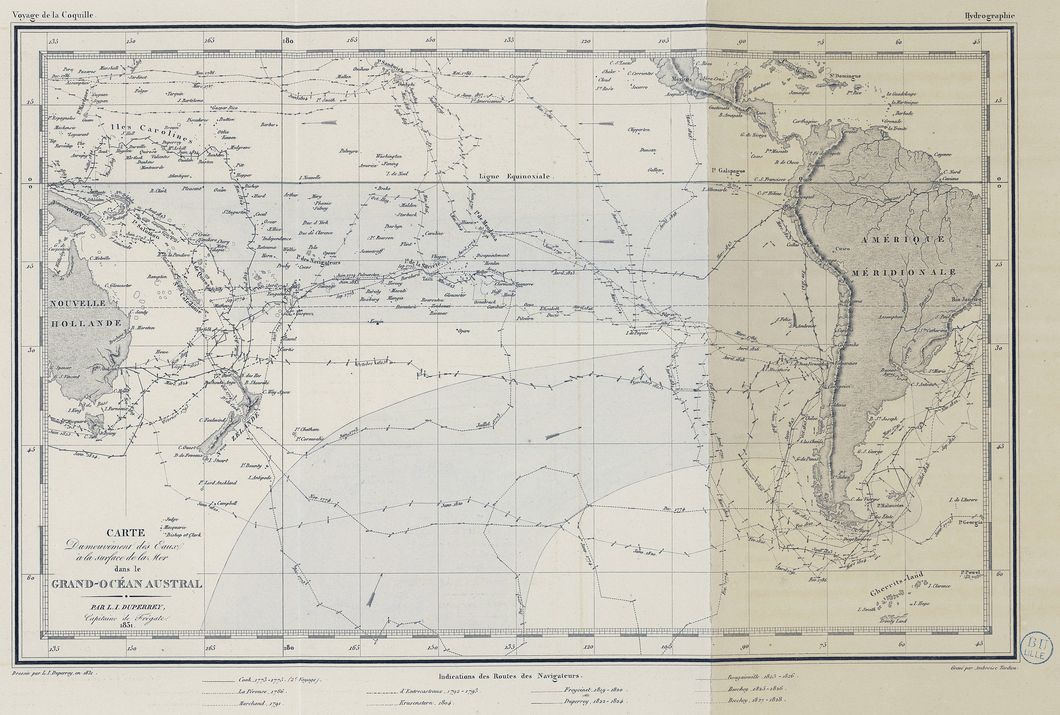
Voyage autour du monde en 1822-1825
Célébrez la Journée mondiale de l’art avec nous !

Coup de théâtre !
Crédit image : Saverio della Gatta, Performers of a Commedia dell'Arte, 1827, WikiCommons .

Projet CoESciTer
LillOnum
5 819 documents disponibles dans LillOnum


Nouveautés

Oeuvres diverses. Tome second
Date : 1756
Sujet : Pensée politique et sociale -- France -- 18e siècle Discours français Morale

Oeuvres diverses. Tome premier
Date : 1756
Sujet : Pensée politique et sociale -- France -- 18e siècle Discours français Morale

D. Severini Boethii... De consolatione philosophiae libri V... Johannis Murmellii (partim etiam Rodolphi Agricolae) commentariis illustrati. Recognovit omnia Johannes Caesarius
Date : 1535
Sujet : Boèce (0480?-0524). De la consolation de la philosophie Philosophie et religion Finalité Philosophie médiévale -- Influence grecque Bonheur (philosophie)


Coustumes locales et particulieres de la ville et bourgeoisie de La Gorgue et de la loy d'Arras, decretees comme s'ensuit
Date : 1752
Sujet : Droit coutumier -- France -- Arras (Pas-de-Calais ; région) -- 18e siècle Droit coutumier -- France -- La Gorgue (Nord) -- 18e siècle

Le porte-feuille d'un homme de goût ou l'esprit de nos meilleurs poëtes. Tome troisième
Date : 1780
Sujet : Poésie française -- Anthologies -- 18e siècle

Le porte-feuille d'un homme de goût ou l'esprit de nos meilleurs poëtes. Tome second
Date : 1780
Sujet : Poésie française -- Anthologies -- 18e siècle

Le porte-feuille d'un homme de goût ou l'esprit de nos meilleurs poëtes. Tome premier
Date : 1780
Sujet : Poésie française -- Anthologies -- 18e siècle


Tabulae de schematibus et tropis. In Rhetorica Philippi Melanchthonis tabulae. In Erasmi Roterdami libellum de duplici copia
Date : 1546
Sujet : Érasme (1469-1536). De duplici copia verborum ac rerum Melanchthon, Philippus (1497-1560). De rhetorica Latin (langue) -- Rhétorique -- Analyse du discours

L'harmonie imitative de la langue française, Poeme en quatre chants
Date : 1788
Sujet : Harmonie (esthétique) -- Poésie française -- 18e siècle Critique littéraire -- France -- 18e siècle

La seconde Sepmaine de Gvillavme de Bartas
Date : 1603
Sujet : Création (religion) -- Poésie française -- 16e siècle